{kind=link}
{kind=link}
{kind=link}
{kind=link}
![]()
Anneri Lötter, Guangyi Chen and Susanne P. Pfeifer - writers
Luis Paulin - coding
Damaris Lattimer - coding
Kyuil Cho - sys. admin, coding
Yilei Fu - coding
Pavel Avdeyev - coding
Wouter De Coster - Team Lead
- To develop a tool to detect and genotype (in terms of length) STR in long reads (de novo) without the need of genome annotation beforehand
- This tool should be applicable in mammals and plants (basically any phased eukaryotic assembly)
Short tandem repeats (STRs) are motifs of multiple nucleotides in length that are repeated. Due to their repetitive nature, the are subject to high mutation rates and cause several genetic diseases including more than 40 neurological and developmental disorders. Using short-read sequencing data to identify and characterize STRs have been met with some shortcomings including biases introduced by use of PCR. Long-read sequencing can be used to identify their length more accurately than short reads as reads can span across the entire repeat region. Long-reads still have a high error rate.
Although tools have been developed to address the high error-rate problem, they still have limitations such as not being able to consider multiple STRs in a single reads. To address these issues, we present STRdust, a tool to (de novo) detect and genotype STRs in long-read sequencing data without prior genome annotation that can be applied in mammals and plants.
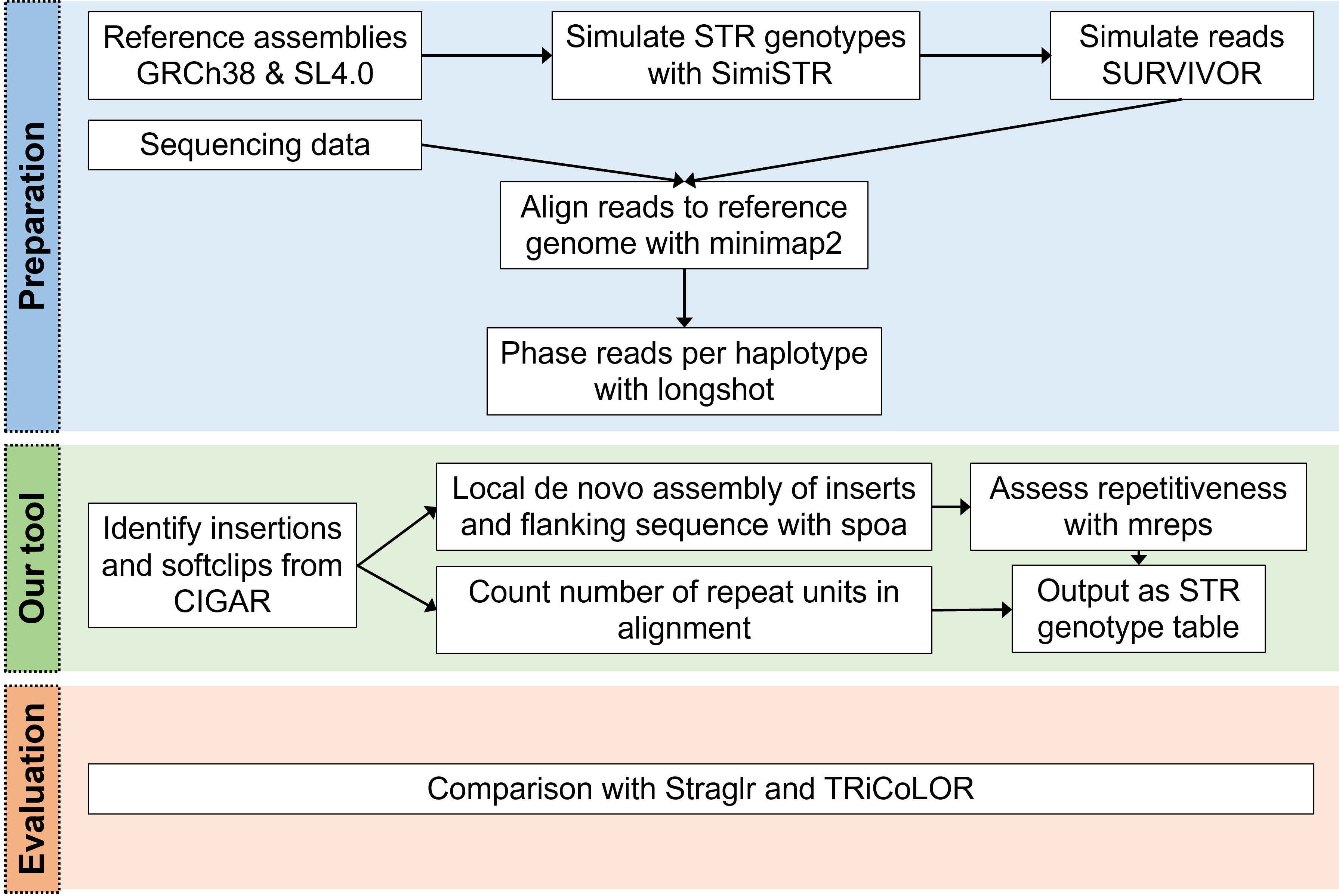
STRdust parses the CIGAR of each read, either genome-wide or in user-specified locus, in order to identify sufficiently large (>15bp) insertions or softclipped bases which could indicate the presence of an enlarged STR. The sequence of those candidate-expansions is extracted, along with 50bp of flanking sequence. Leveraging the phased input data, such insertions are combined per haplotype when multiple of these are found closeby (within 50bp) across multiple reads. The combination is done using spoa, which generates a multiple sequence alignment and from that a consensus sequence. The obtained consensus sequence, in which inaccuracies inherent to the long read sequencing technologies should be reduced, is then used in mreps, which will assess the repetitive character of the sequence and isentify the repeat unit.
-
Clone git repository
git clone https://github.com/collaborativebioinformatics/STRdust.git -
Change to git repository folder
cd <repository-folder-path> -
Install environment
conda create --name STRdust -c bioconda python=3.7 pysam spoa pyspoa mreps -
Activate environment
conda activate STRdust -
Dry run
python STRdust/STRdust.py -h
pip install setup.py
STRdust includes some third-party software:
python STRdust/STRdust.py path_to_bamfile
usage: Genotype STRs from long reads [-h] [-o OUT_DIR] [-d DISTANCE]
[-r MREPS_RES] [--save_temp] [--debug]
[--region REGION]
bam
positional arguments:
bam phased bam file
optional arguments:
-h, --help show this help message and exit
-o OUT_DIR, --out_dir OUT_DIR
output directory (tool directory by default)
-d DISTANCE, --distance DISTANCE
distance across which two events should be merged
-r MREPS_RES, --mreps_res MREPS_RES
tolerent error rate in mreps repeat finding
--save_temp enable saving temporary files in output directory
--debug enable debug output
--region REGION run on a specific interval only
cd STRdust
python STRdust/STRDust.py test_data/subsampled.bam -o test_results
- Phased bam alignment file
- a table with STR genotype calls (chromosome, start, end, repeat motif and repeat size)
| #chrom | start | end | repeat_seq | size |
|---|---|---|---|---|
| 22 | 10521893 | 10521909 | GCA | 16 |
| 22 | 10522628 | 10522647 | AATA | 19 |
| 22 | 10534566 | 10534586 | GTTTT | 20 |
| 22 | 10511934 | 10511957 | AG | 23 |
| 22 | 10516550 | 10516595 | TA | 45 |
| 22 | 10516550 | 10516572 | TATATATG | 22 |
SimiSTR was used to modify the GRCh38 (human) and SL4.0 (tomato) reference genome assemblies. Then, additional variation (SNVs) were introduced with SURVIVOR at a rate of 0.001.
Long reads were simulated using SURVIVOR for the GRCh38 (human) and SL4.0 (tomato) STR-modified genomes. Mapping was performed with minimap2 two-fold (with and without the -Y parameter), and phasing was done with longshot. Default parameters were used for all tools, if not otherwise mentioned.
We made use of SimiSTR, which manipulates a reference file (genome, chromosome) in order to simulate STR. The simulator takes a haploid file as reference(.fasta) and a region file (.bed) containing information about known STR-regions as input. All of the supplied regions can be, expanded and mutated. The output result is a fasta file with the modified STRs, and can be haploid or diploid (homozygosity can be user defined).
This tool was tested using simulated reads for human chromosome 22 and tomato chromosome 1. Long reads were simulated using SimiSTR for the GRCh38 (human) and SL4.0 (tomato) reference genome assemblies. The simulator takes a haploid file as reference (.fasta) and a region file (.bed) containing information about known STR-regions as input. All of the supplied regions can be modified in
expansion (% of regions that will randomly be positive or negative expanded [0.00-1.00]),
mutation (% chance for a base to be substituted [0.00-1.00]),
number of indels (X times less likely than chance for mutation to insert or delete a base [0.00-1.00]). Further can the simulation file (.fasta) be created as
haploid [h] or
diploid [d]. If diploid is chosen,
the percentage of regions that should get homozygous can be set [0.00-1.00].
The simulator works on assembled genomes, as well as on only one or more assembled chromosomes, if the bed-file contains such entrances likewise (anything else could run error-free, but will not manipulate anything, as manipulations only occur in the known regions). The simulated reads were then used to create a phased bam that was used as input for STRdust.
We compared STRdust to Straglr and TRiCoLOR. Both tools used as input the phased alignment, and were used with default parameters.